





PARAM KILIMANJARO SUPERCOMPUTER (HPC) INFRASTRUCTURE
Super Computer Param Kilimanjaro (HPC) is a Linux Based Open Source PARAM Supercomputing Cluster which consisting of 13 - Compute Nodes with the highly computing power of 14 Tera Flops. The HPC facility contains the following; 1 - Master Node, 6 - Compute Nodes, 1-Compute Node with GPGPU Accelerator, 2- Storage Nodes, 1 - Storage Expansion, 1-Backup Node and 1-Tape Library. The HPC also includes SAS Based Storage of 100 TB of storage capacity with Interconnects of low latency high-speed network which include Infiniband, Gigabit and Ethernet high speed Network Switches.
PARAM Kilimanjaro HPC Cluster Hardware Mapping:
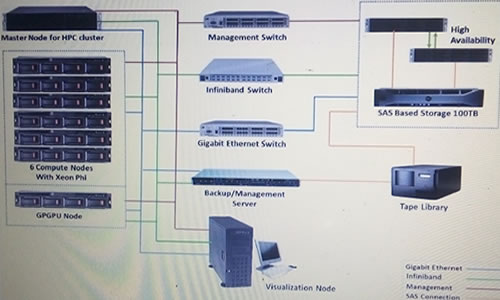
HPC Cluster Specifications:

| 1 * Master Node: Dell Power Edge R 730 | 6 * Compute Nodes: Dell Power Edge R730 with Intel Xeon Phi-coprocessor | 1 * Compute Node: Dell Power Edge R 730 with GPGPU Accelerator | 2 * Storage Nodes: Dell Power Edge R 730 |
|---|---|---|---|
|
|
|
|
| 1 * Storage Expansion : MD 3060 e (100 TB Usable) | 1 * Backup Node : Dell PowerEdge R 730 | 1 * Tape Library : TL 2000 |
|---|---|---|
|
|
|
Software Specifications:
| Operating System | Cent OS Linux OS v. 7.2 |
|---|---|
|
|
Data Analysis Software Tools Implemented in PARAM Kilimanjaro HPC
| Bioinformatics Applications and Molecular Modelling | GPGPU Applications | Climate and Ocean Modelling | Data Visualization Tools |
|---|---|---|---|
|
|
|
|
Uses of some PARAM HPC Tools:-
1. Bio-informatics and Molecular Dynamics Applications
Using NAMD Tool in HPC, we can perform parallel molecular dynamics code design for high performance simulation of a large bio-molecular system. NAMD works well with CHARMM potential functions, parameters and file formation. NAMD only uses the GPU for non-bonded force evaluation. Energy evaluation is done on the GPU.
2. GROMACS
In HPC GROMACS is a versatile and extremely well optimized package to perform molecular dynamics computer simulations and subsequent trajectory analysis.
In its development for biochemical molecules like proteins, lipids, and nucleic acids that have a lot of complicated bonded interactions GROMACS provides a complete modelling package in HPC for protein , membranes systems etc, it includes mode analysis, essential dynamics analysis and many trajectory analysis utilities.
3. GPU - HMMer: GPU - HMMer is a protein sequence analysis tool which used to predict homology structure and function of particular peptide sequences exist in abundance. It allows constructing profile Hidden Markov Models(HMM's) of a set of aligned protein sequences with known similar function and homology, and provides database search functionality to compare input HMMs to sequence databases.
HMMer's hmmsearch tool is particularly well-suited for many core architectures due to the embarrassingly parallel nature of sequence database searches, so in GPU -HMMer MRF is parallelized and P7Vterbi Kernel is implemented which is a C Code of the P7Vterbi algorithm which is gpu enabled ported on CUDA with a variety of performance optimization.
MPQC (Massively Parallel Quantum Chemistry);
Is the massively Parallel Quantum Chemistry Program. It computes properties of atoms and molecules from first Principles using the time independent Schrodinger Equation. It runs on a wide range of architectures ranging from individual workstations to sysmmetric multiprocessors to massively parallell computers. Its design is object oriented using the C++ Programming Language.
ABINIT ;
Is a package whose main program allows one to find the total energy, change density and electronic structure of systems made of electrons and nuclei (molecules and periodic solids) within Density Functional Theory(DFT), using pseudo potentials and a plane wave basis. Excited states can be computed within the Time-Dependent Density Functional Theory(for molecules), or within Many-Body Perturbation Theoty (the GW approximation).
CUDA MEME;
Is a tool for dicovering motifs in a group of related DNA or protein sequences. A motif is a sequennce pattern that occurs repeatedly in a group of related protein or DNA Sequences. MEME represents motifs as position -dependent latter -probability matrices which describe the probability of each possible letter at each position in the pattern.
MUMmer-GPU ;
Is a bio-informatics software for high-throughput sequence alignment using GPUs. It accelerates the alignment of multiple query sequences against a single refence sequence by taking advantage of the massively parallel processing CUDA architecture of NVIDIA Tesla GPUs.
Weather Research and Forecasting (WRF)
WRF Model Is a next-generation mesoscale nmerical weather prediction system designed to serve both operational forecasting and atmospheric research needs. WRF is suitable for a broad spectrum of application across scales ranging from meters to thousands of kilometers .
WRF software has been designed for performance portability, maintainability, extensibility, readability, usability, run-time configurability, interoperability, and reuse in a limited area model which later boundary conditions and nesting.
RegCM Model ;
RegCM Model is a 3-dimensional, sigma-coordinate, primitive equation regional climate model.
The model is compressible based on primitive equations and employs a terrain following r-vertical coordinat. The model includes parameterizations of a surface boundary layer and moist processes which account for the physical exchanges between the land and surface boundary layer and free atmosphere. Furthermore, the RegCM3 has options to inteface with a variety of reanalysis and General Circulation Model (GCM) boundary conditions.
ROMS (Regional Ocean Modeling System) Model ;
ROMS is a free-surface terrain following primitive equations ocean model widely used by the scientific community for a diverse range of applications. ROMS includes accurate and efficient physical and numerical algorithms and several coupled models for biogeochemical, bio-optical sediment and sea ice applications. It also includes several vertical mixing schemes multiple levels of nesting and composed grids.
mpiBLAST;
Basic Local Alignment Search Tool (BLAST) finds regions of similarity between biological sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance. mpiBLAST is a freely available open source, parallel implementation of NCBI BLAST.
By efficintly utilizing distributed computational resources rthrough database fragmentation, queryu segmentation, intelligent scheduling, and parallel I/O, mpiBLAST improves NCBI BLAST performance by several orders of magintude while scaling to hundreads of processors. mpiBLAST is also portable across many different platforms and operating systems.
CGView (Circular Genome Viewer);
CGView is a Java package for generating high quality, zoomable maps of circular genomes. Its primary purpose is to serve as a component of sequence annotation pipelines, as a means of generating visual output suitable for the web. Feature information and rendering options are supplied to the program using an XML file, a tab delimited file, or an NCBI ptt file. CGView converts the input into a graphical map(PNG, JPG, or Scalable Vector Graphical formats), complete with labels, a title. legends, and footnotes. In addition to the default full view map, the program can generate a series of hyperlinked maps showing expanded views. The linked maps can be explored using any web browser, allowing rapid genome browsing, and facilitating data sharing. The feature labels in maps an be hyperlinked to external resources, allowing CGView maps to be integrated with existing website content or databases.





